IVD ĶŻĮÕōüŃü»ŃĆüÕüźÕ║ĘńŖȵģŗŃü«Ķ©║µ¢ŁŃĆüµ▓╗ńÖéŃĆüŃüŠŃü¤Ńü»õ║łķś▓Ńü½õĮ┐ńö©ŃüĢŃéīŃéŗŃāćŃāÉŃéżŃé╣ŃüŖŃéłŃü│ŃéĘŃé╣ŃāåŃāĀŃü¦ŃüÖŃĆéŃüØŃéīŃéēŃü»ŃĆüĶĪƵČ▓ŃĆüÕöŠµČ▓ŃĆüńĄäń╣öŃü¬Ńü®Ńü«ńö¤ńē®ÕŁ”ńÜäŃéĄŃā│ŃāŚŃā½Ńü«ÕÅÄķøåŃü©µż£µ¤╗Ńü½õĮ┐ńö©ŃüĢŃéīŃéŗŃüōŃü©ŃéƵäÅÕø│ŃüŚŃü”ŃüäŃüŠŃüÖŃĆéŃéĄŃā│ŃāŚŃā½Ńü»ŃĆüķ╝╗Ńü«ÕåģÕü┤ŃéäÕ¢ēŃü«ÕźźŃüŗŃéēŃĆüŃüŠŃü¤Ńü»ķØÖĶäłŃéäµīćÕģłŃüŗŃéēµÄĪÕÅ¢Ńü¦ŃüŹŃüŠŃüÖŃĆé
IVD ĶŻĮÕōüŃü»ķØ×õŠĄĶź▓ńÜäŃü¦ŃüéŃéŗŃüōŃü©Ńü½µ│©µäÅŃüÖŃéŗŃüōŃü©ŃüīķćŹĶ”üŃü¦ŃüÖŃĆéŃü¤Ńü©ŃüłŃü░ŃĆü Poclight HSCL-5000 Micro ŃāøŃāóŃéĖŃāŗŃéóŃé╣Õī¢ÕŁ”ńÖ║ÕģēŃéżŃāĀŃāÄŃéóŃāāŃé╗Ńéż ŃéóŃāŖŃā®ŃéżŃéČŃā╝ Ńü¦ŃüÖŃĆé
Ńü¤ŃüĀŃüŚŃĆüŃüØŃéīŃéēŃü»õŠØńäČŃü©ŃüŚŃü”Õī╗ńÖéµ®¤ÕÖ©Ńü©Ķ”ŗŃü¬ŃüĢŃéīŃéŗŃü¤ŃéüŃĆüÕÉīŃüśĶ”ÅÕłČŃü«Õ»ŠĶ▒ĪŃü©Ńü¬ŃéŖŃüŠŃüÖŃĆéÕī╗ńÖéµ®¤ÕÖ©Ńü»ŃĆüõ╗źõĖŗŃü«ńø«ńÜäŃü¦õ║║ķ¢ōŃüīõĮ┐ńö©ŃüÖŃéŗŃüōŃü©ŃéƵäÅÕø│ŃüŚŃü¤ŃüéŃéēŃéåŃéŗÕÖ©ÕģĘŃĆüÕÖ©ÕģĘŃĆüŃéĮŃāĢŃāłŃé”Ńé¦ŃéóŃĆüŃüŠŃü¤Ńü»ķ¢óķĆŻµ®¤ÕÖ©Ńü¦µ¦ŗµłÉŃüĢŃéīŃü”ŃüäŃüŠŃüÖŃĆé
┬Ę ńŚģµ░ŚŃéäŃüæŃüīŃü«Ķ©║µ¢ŁŃĆüõ║łķś▓ŃĆüńøŻĶ”¢ŃĆüŃüŠŃü¤Ńü»µ▓╗ńÖéŃĆé
┬Ę ńö¤ÕæĮŃü«ńČŁµīüŃüŠŃü¤Ńü»ńČŁµīüŃĆé
┬Ę ÕÅŚĶāÄŃü«Ķ¬┐ń»ĆŃĆé
Ńā╗ Õī╗ńÖéµ®¤ÕÖ©Ńü«µČłµ»ÆŃĆé
Ńā╗ ŃāÆŃāłŃüŗŃéēńö¤õĮōĶ®”µ¢ÖŃéƵÄĪÕÅ¢ŃüŚŃĆüÕī╗ńÖéńø«ńÜäŃü¦µāģÕĀ▒ŃéƵÅÉõŠøŃüÖŃéŗŃĆé
In Vitro Diagnostics Ńü«ĶŻĮķĆĀµźŁĶĆģŃü»ŃĆüÕÅéÕģźŃüŚŃü¤ŃüäÕĖéÕĀ┤Ńü½Õ┐£ŃüśŃü”ŃĆüµ¼¦ÕĘ×ķĆŻÕÉłŃĆüĶŗ▒ÕøĮŃĆüŃüŖŃéłŃü│ń▒│ÕøĮŃü«Ķ”ÅÕłČĶ”üõ╗ČŃéƵ║ĆŃü¤ŃüÖÕ┐ģĶ”üŃüīŃüéŃéŖŃüŠŃüÖŃĆé
ŃüōŃéīŃéēŃü«Ķ”üõ╗ČŃü½Ńü»ŃĆüEU Ńü« In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746 ŃüŖŃéłŃü│Ķŗ▒ÕøĮŃü« UK Medical Devices Regulations (UK MDR) 2002 ŃüīÕɽŃüŠŃéīŃüŠŃüÖŃĆéń▒│ÕøĮŃü¦Ńü»ŃĆüIVD ĶŻĮÕōüŃü»ŃĆüķĆŻķé”ķŻ¤ÕōüÕī╗Ķ¢¼ÕōüÕī¢ń▓¦Õōüµ│Ģ (FD&C µ│Ģ) Ńü«Ńé╗Ńé»ŃéĘŃā¦Ńā│ 201(h) Ńü¦Õ«ÜńŠ®ŃüĢŃéīŃü”ŃüäŃüŠŃüÖŃĆéŃüōŃéīŃéēŃü«ŃāćŃāÉŃéżŃé╣Ńü»ŃĆüÕģ¼ĶĪåĶĪøńö¤ŃéĄŃā╝ŃāōŃé╣µ│Ģń¼¼ 351 µØĪŃĆüŃüŖŃéłŃü│ 1988 Õ╣┤Ńü«Ķć©Õ║Ŗµż£µ¤╗µö╣Õ¢äõ┐«µŁŻµØĪķĀģ (CLIA) Ńü½Õ¤║ŃüźŃüÅÕłåķĪ×Ńü«Õ»ŠĶ▒ĪŃü©Ńü¬ŃéŗÕĀ┤ÕÉłŃééŃüéŃéŖŃüŠŃüÖŃĆé
IVDR Ńü½ŃéłŃéŗŃü©ŃĆüIVD ĶŻĮÕōüŃü»ŃĆüŃĆīĶ®”Ķ¢¼ŃĆüĶ®”Ķ¢¼ĶŻĮÕōüŃĆüŃéŁŃāŻŃā¬Ńā¢Ńā¼Ńā╝Ńé┐ŃĆüŃé│Ńā│ŃāłŃāŁŃā╝Ńā½µØɵ¢ÖŃĆüŃéŁŃāāŃāłŃĆüµ®¤ÕÖ©ŃĆüĶŻģńĮ«ŃĆüµ®¤ÕÖ©ŃĆüŃéĮŃāĢŃāłŃé”Ńé¦ŃéóŃĆüŃüŠŃü¤Ńü»ŃéĘŃé╣ŃāåŃāĀŃü¦ŃüéŃéŖŃĆüÕŹśńŗ¼Ńü¦õĮ┐ńö©ŃüĢŃéīŃéŗŃüŗńĄäŃü┐ÕÉłŃéÅŃüøŃü”õĮ┐ńö©ŌĆŗŌĆŗŃüĢŃéīŃéŗŃüŗŃéÆÕĢÅŃéÅŃüÜŃĆüµäÅÕø│ŃüĢŃéīŃü¤ŃüéŃéēŃéåŃéŗÕī╗ńÖéµ®¤ÕÖ©ŃĆŹŃü©Õ«ÜńŠ®ŃüĢŃéīŃü”ŃüäŃüŠŃüÖŃĆéõ║║õĮōŃü½ńö▒µØźŃüÖŃéŗĶĪƵČ▓ŃéäńĄäń╣öŃü«µÅÉõŠøŃéÆÕɽŃéƵż£õĮōŃü«µż£µ¤╗Ńü«Ńü¤ŃéüŃü½ŃĆüĶŻĮķĆĀµźŁĶĆģŃü½ŃéłŃüŻŃü” in vitro Ńü¦õĮ┐ńö©ŃüĢŃéīŃéŗŌĆ”ŃĆŹ
Õ░éķ¢ĆÕ«ČŃü»ŃĆüŃüÖŃü╣Ńü”Ńü«Ķć©Õ║Ŗµ▒║Õ«ÜŃü«ń┤ä 70% Ńüī IVD ĶŻĮÕōüŃéÆõĮ┐ńö©ŃüŚŃü”ĶĪīŃéÅŃéīŃü”ŃüäŃéŗŃü©µÄ©Õ«ÜŃüŚŃü”ŃüäŃüŠŃüÖŃĆé
Õ”ŖÕ©Āµż£µ¤╗ŃĆüCOVID-19 µż£µ¤╗ŃĆüŃüŖŃéłŃü│ HIV µż£µ¤╗Ńü»ŃĆüIVD ĶŻĮÕōüŃü«õŠŗŃü¦ŃüÖŃĆéPoclight HSCL-5000 Micro CLIA Analyzer IVD ŃāćŃāÉŃéżŃé╣Ńü«ŃüØŃü«õ╗¢Ńü«õŠŗŃü©ŃüŚŃü”Ńü»ŃĆüµ¼ĪŃü«ŃééŃü«ŃüīŃüéŃéŖŃüŠŃüÖŃĆé
Ńā╗ ŃüīŃéōĶ©║µ¢Ł
Ńā╗ńö▓ńŖČĶģ║Ķ©║µ¢Ł
Ńā╗ńéÄńŚćĶ©║µ¢Ł
┬Ę ŃéżŃāĀŃāÄŃéóŃāāŃé╗Ńéż
┬Ę ...
Õī╗ńÖ鵿£õĮōńö©Ńü½ĶŻĮķĆĀŃüĢŃéīŃü¤Ńā¼Ńé╗ŃāŚŃé┐Ńé»Ńā½ŃééIVDĶŻĮÕōüŃü¦ŃüÖŃĆéõĖ¢ńĢīõ┐ØÕüźµ®¤ķ¢óŃü½ŃéłŃéŗŃü©ŃĆüńÅŠÕ£© 40,000 ŃéÆĶČģŃüłŃéŗĶŻĮÕōüŃüī IVD µż£µ¤╗Ńü½Õł®ńö©Ńü¦ŃüŹŃüŠŃüÖŃĆéŃüōŃéīŃéēŃü»ŃĆüÕŠōµØźŃü«Ķć©Õ║Ŗµż£µ¤╗ŃüŗŃéēŃāØŃéżŃā│ŃāłŃé¬Ńā¢Ńé▒Ńéóµż£µ¤╗ŃüŠŃü¦ŃüĢŃüŠŃü¢ŃüŠŃü¦ŃüÖŃĆé
ń▒│ÕøĮŃü¦Ńü»ŃĆüFDA Ńü» IVD ĶŻĮÕōüŃéÆÕɽŃéĆŃüÖŃü╣Ńü”Ńü«Õī╗ńÖéµ®¤ÕÖ©ŃéÆŃé»Ńā®Ńé╣ IŃĆüŃé»Ńā®Ńé╣ IIŃĆüŃüŠŃü¤Ńü»Ńé»Ńā®Ńé╣ III Ńü½ÕłåķĪ×ŃüŚŃü”ŃüäŃüŠŃüÖŃĆéŃāćŃāÉŃéżŃé╣Ńü«ÕłåķĪ×Ńü»ŃĆüķ¢óķĆŻŃüÖŃéŗŃā¬Ńé╣Ńé»Ńü©ŃĆüĶŻĮÕōüŃü«Õ«ēÕģ©µĆ¦ŃéÆõ┐ØĶ©╝ŃüÖŃéŗŃü¤ŃéüŃü½Õ┐ģĶ”üŃü¬Ķ”ÅÕłČń«ĪńÉåŃü«Ńā¼ŃāÖŃā½Ńü½Õ¤║ŃüźŃüäŃü”ńĢ░Ńü¬ŃéŖŃüŠŃüÖŃĆéŃüŚŃü¤ŃüīŃüŻŃü”ŃĆüIVDÕłåķĪ×Ńü»ŃĆüŃāĪŃā╝Ńé½Ńā╝ŃüīĶŻĮÕōüŃéÆÕĖéÕĀ┤Ńü½µŖĢÕģźŃüÖŃéŗŃü¤ŃéüŃü½ÕŠōŃüåÕ┐ģĶ”üŃüīŃüéŃéŗÕĖéĶ▓®ÕēŹŃāŚŃāŁŃé╗Ńé╣ŃéƵ▒║Õ«ÜŃüŚŃüŠŃüÖŃĆé
┬Ę Ńé»Ńā®Ńé╣ I ĶŻĮÕōüŃü»ŃĆüõĮÄŃüŗŃéēõĖŁń©ŗÕ║”Ńü«Ńā¬Ńé╣Ńé»Ńü©Ķ”ŗŃü¬ŃüĢŃéīŃĆüõĖĆĶł¼ńÜäŃü¬ń«ĪńÉåŃüīÕ┐ģĶ”üŃü¦ŃüÖŃĆé
┬Ę Ńé»Ńā®Ńé╣ II ĶŻĮÕōüŃü»õĖŁń©ŗÕ║”ŃüŗŃéēķ½śŃā¬Ńé╣Ńé»Ńü©Ķ”ŗŃü¬ŃüĢŃéīŃĆüõĖĆĶł¼ńÜäŃü¬ń«ĪńÉåŃü©ńē╣ÕłźŃü¬ń«ĪńÉåŃüīÕ┐ģĶ”üŃü¦ŃüÖŃĆé
┬Ę Ńé»Ńā®Ńé╣ III ĶŻĮÕōüŃü»Ńā¬Ńé╣Ńé»Ńüīķ½śŃüäŃü©Ķ”ŗŃü¬ŃüĢŃéīŃĆüõĖĆĶł¼ń«ĪńÉåŃü©ÕĖéĶ▓®ÕēŹµē┐Ķ¬Ź (PMA) ŃüīÕ┐ģĶ”üŃü¦ŃüÖŃĆé
ŃāćŃāÉŃéżŃé╣ Ńé»Ńā®Ńé╣ŃüīÕóŚÕŖĀŃüÖŃéŗŃü½ŃüżŃéīŃü”ŃĆüĶ”ÅÕłČń«ĪńÉåŃééÕóŚÕŖĀŃüÖŃéŗŃüōŃü©Ńü½µ│©µäÅŃüÖŃéŗŃüōŃü©ŃüīķćŹĶ”üŃü¦ŃüÖŃĆéŃé»Ńā®Ńé╣ I IVR ŃāćŃāÉŃéżŃé╣Ńü»Ķ”ÅÕłČń«ĪńÉåŃüīµ£ĆŃééńĘ®ŃüäŃü«Ńü½Õ»ŠŃüŚŃĆüŃé»Ńā®Ńé╣ III IVR ŃāćŃāÉŃéżŃé╣Ńü»µ£ĆŃééÕÄ│ŃüŚŃüäĶ”üõ╗ČŃéÆÕéÖŃüłŃü”ŃüäŃüŠŃüÖŃĆéń▒│ÕøĮŃü¦Ńü«õĮōÕż¢Ķ©║µ¢ŁĶŻĮÕōüŃü«ÕĖéÕĀ┤µŖĢÕģźŃéƵż£Ķ©ÄŃüŚŃü”ŃüäŃéŗŃā®ŃéżŃāĢ ŃéĄŃéżŃé©Ńā│Ńé╣õ╝üµźŁŃü»ŃĆüFDA Ńü«ŃĆīClassify Your Medical DeviceŃĆŹŃā¬ŃéĮŃā╝Ńé╣ŃéÆÕÅéńģ¦Ńü¦ŃüŹŃüŠŃüÖŃĆé
µ¼¦ÕĘ×ķĆŻÕÉłŃü¦Ńü«Ķ”ÅÕłČķüĄÕ«łŃéÆńó║õ┐ØŃüŚŃü¤ŃüäŃü¦ŃüÖŃüŗ? IVDR Ńü» 2021 Õ╣┤Ńü½ŃéóŃāāŃāŚŃé░Ńā¼Ńā╝ŃāēŃüĢŃéīŃĆüµ¼ĪŃü«ŃéłŃüåŃü¬µö╣Õ¢äŃüīĶĪīŃéÅŃéīŃüŠŃüŚŃü¤ŃĆé
┬Ę ŃāćŃāÉŃéżŃé╣Ńü«ŃāłŃā¼Ńā╝ŃéĄŃāōŃā¬ŃāåŃéŻŃĆüńÖ╗ķī▓ŃĆüŃüŖŃéłŃü│ķü®ÕłćŃü¬Ńā®ŃāÖŃā½ĶĪ©ńż║Ńü«µż£Ķ©╝Ńü½ķ¢óŃüÖŃéŗĶŻĮķĆĀµźŁĶĆģŃĆüĶ╝ĖÕģźµźŁĶĆģŃĆüŃüŖŃéłŃü│Ķ▓®ÕŻ▓µźŁĶĆģŃü«µśÄńó║Ńü¬ńŠ®ÕŗÖŃĆéŃüōŃéīŃü½Ńü»ŃĆüõĖƵäÅŃü«ŃāćŃāÉŃéżŃé╣ĶŁśÕłźÕŁÉ (UDI) Ńü½Õ¤║ŃüźŃüäŃü¤ŃāłŃā¼Ńā╝ŃéĄŃāōŃā¬ŃāåŃéŻ ŃéĘŃé╣ŃāåŃāĀŃüīÕɽŃüŠŃéīŃüŠŃüÖŃĆé
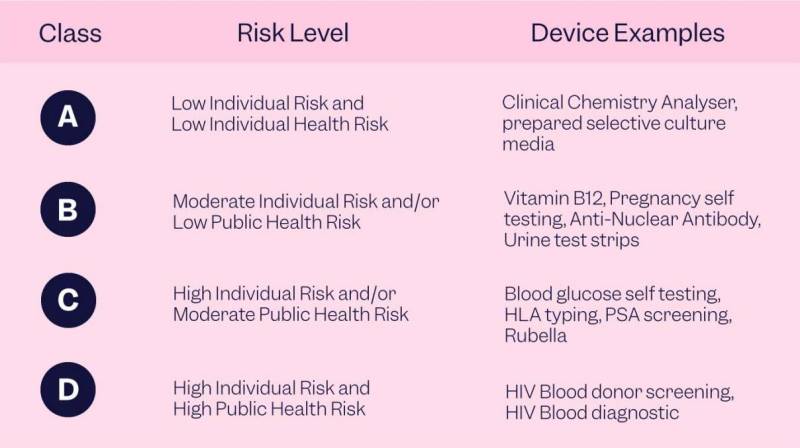
┬Ę 4 ŃüżŃü«Ńā¬Ńé╣Ńé» Ńé»Ńā®Ńé╣Ńü¦µ¦ŗµłÉŃüĢŃéīŃéŗÕłåķĪ×Ńü«Õ░ÄÕģźŃĆéŃé»Ńā®Ńé╣ A ŃāćŃāÉŃéżŃé╣Ńü»ŃĆüÕĆŗõ║║ŃüŖŃéłŃü│Õģ¼ĶĪåŃü«ÕüźÕ║ĘŃā¬Ńé╣Ńé»ŃüīõĮÄŃüäŃüōŃü©ŃéÆńē╣ÕŠ┤Ńü©ŃüŚŃü”ŃüŖŃéŖŃĆüŃé»Ńā®Ńé╣ B ŃāćŃāÉŃéżŃé╣Ńü»ŃĆüÕĆŗõ║║ŃüŖŃéłŃü│/ŃüŠŃü¤Ńü»Õģ¼ĶĪåĶĪøńö¤Ńā¬Ńé╣Ńé»ŃüīõĖŁń©ŗÕ║”Ńü¦ŃüéŃéŗŃüōŃü©ŃéÆńē╣ÕŠ┤Ńü©ŃüŚŃü”ŃüŖŃéŖŃĆüŃé»Ńā®Ńé╣ C ŃāćŃāÉŃéżŃé╣Ńü»ŃĆüÕĆŗõ║║Ńā¬Ńé╣Ńé»Ńüīķ½śŃüÅŃĆüŃüŖŃéłŃü│/ŃüŠŃü¤Ńü»Õģ¼ĶĪåĶĪøńö¤Ńā¬Ńé╣Ńé»ŃüīõĖŁń©ŗÕ║”Ńü¦ŃüéŃéŗŃüōŃü©ŃéÆńē╣ÕŠ┤Ńü©ŃüŚŃü”ŃüŖŃéŖŃĆüŃé»Ńā®Ńé╣ D ŃāćŃāÉŃéżŃé╣Ńü»ŃĆüÕĆŗõ║║Ńā¬Ńé╣Ńé»Ńüīķ½śŃüÅŃĆüŃüŗŃüż/ŃüŠŃü¤Ńü»Õģ¼ĶĪåĶĪøńö¤Ńā¬Ńé╣Ńé»Ńüīķ½śŃüäŃüōŃü©ŃéÆńē╣ÕŠ┤Ńü©ŃüŚŃü”ŃüäŃéŗŃĆéÕģ¼ĶĪåĶĪøńö¤õĖŖŃü«Ńā¬Ńé╣Ńé»ŃĆé
┬Ę Ńā¬Ńé╣Ńé»Ńü«ķ½śŃüä IVD µ®¤ÕÖ©Ńü«ŃéłŃéŖÕÄ│µĀ╝Ńü¬ń«ĪńÉåŃĆé
┬Ę ńē╣Õ«ÜŃü«õĖĆķĆŻŃü«Õ¤║µ║¢ŃéÆÕ╝ĘÕī¢ŃüÖŃéŗŃüōŃü©Ńü½ŃéłŃéŖŃĆüķĆÜń¤źµ®¤ķ¢ó (ŃüŠŃü¤Ńü»ńŗ¼ń½ŗŃüŚŃü¤ń¼¼õĖēĶĆģķü®ÕÉłµĆ¦Ķ®ĢõŠĪµ®¤ķ¢ó) Ńü«ńøŻńØŻŃéÆõ┐ØĶ©╝ŃüŚŃüŠŃüÖŃĆé
┬Ę EU Ńü«Õī╗ńÖéµ®¤ÕÖ©Ńü½ķ¢óŃüÖŃéŗÕīģµŗ¼ńÜäŃü¬ŃāćŃā╝Ńé┐ŃāÖŃā╝Ńé╣ (EUDAMED Ńü©Õæ╝Ńü░ŃéīŃéŗ) Ńü½ŃéłŃéŗķĆŵśÄµĆ¦Ńü«ÕÉæõĖŖŃĆé
┬Ę ńē╣Ńü½ÕĖéÕĀ┤ńøŻĶ”¢Ńüīķ¢óõ┐éŃüÖŃéŗÕĀ┤ÕÉłŃĆüEU Ķ½ĖÕøĮķ¢ōŃü«ŃéłŃéŖÕżÜŃüÅŃü«Ķ¬┐µĢ┤ŃéÆõ╝┤ŃüåŃĆüĶć©Õ║ŖńÜäĶ©╝µŗĀŃü©ŃāæŃāĢŃé®Ńā╝Ńā×Ńā│Ńé╣Ķ®ĢõŠĪĶ”ÅÕēćŃü«Õ╝ĘÕī¢ŃĆé
┬Ę ŃāĪŃā╝Ńé½Ńā╝Ńü«ÕĖéĶ▓®ÕŠīńøŻĶ”¢Ķ”üõ╗ČŃü«Õ╝ĘÕī¢ŃĆé
┬Ę ÕÉīŃüśµ®¤ķ¢óÕåģŃü¦ĶŻĮķĆĀŃüŖŃéłŃü│õĮ┐ńö©ŃüĢŃéīŃéŗŃĆīńżŠÕåģµ®¤ÕÖ©ŃĆŹŃüŠŃü¤Ńü» IVD ĶŻĮÕōüŃü½ķ¢óŃüÖŃéŗńē╣Õ«ÜŃü«Ķ”üõ╗ČŃĆé
IVD ĶŻĮÕōüŃéÆÕ«ēÕģ©ŃüŗŃüżÕŖ╣µ×£ńÜäŃü½ÕĖéÕĀ┤Ńü½µŖĢÕģźŃüÖŃéŗŃüōŃü©ŃüīķćŹĶ”üŃü¦ŃüéŃéŖŃĆüĶ”ÅÕłČŃéÆńÉåĶ¦ŻŃüÖŃéŗŃüōŃü©ŃüīńÉåµā│ńÜäŃü¬ń¼¼õĖƵŁ®Ńü¦ŃüÖŃĆé
IVD ĶŻĮÕōüŃü»ŃĆüŃüĢŃüŠŃü¢ŃüŠŃü¬ÕüźÕ║ĘńŖȵģŗŃü½Õ»ŠŃüÖŃéŗķćŹĶ”üŃü¬ķś▓ĶĪøńĘÜŃü¦ŃüÖŃĆéIn Vitro Diagnostics Ńü«Õ«ēÕģ©µĆ¦Ńü©ÕōüĶ│¬Ńü»õĖŹÕÅ»µ¼ĀŃü¦ŃüÖŃĆéŃüōŃéīŃüōŃüØŃüīŃĆüĶŻĮķĆĀµźŁĶĆģŃü«Ķ”ÅÕłČķĀåÕ«łŃüīķØ×ÕĖĖŃü½ķćŹĶ”üŃü¦ŃüéŃéŗńÉåńö▒Ńü¦ŃüÖŃĆéPoclight Ńü» CE ŃüŖŃéłŃü│ ISO Ńü«Ķ¬ŹÕ«ÜŃéÆÕÅŚŃüæŃü”ŃüŖŃéŖŃĆüĶ®”ķ©ōŃéÆÕ«ēÕ┐āŃüŚŃü”ĶĪīŃüłŃéŗŃéłŃüåŃü½Ķ®”Ķ¢¼ŃéÆĶ▒ŖÕ»īŃü½ÕÅ¢ŃéŖµÅāŃüłŃü”ŃüäŃüŠŃüÖŃĆé






 IPv6ŃāŹŃāāŃāłŃā»Ńā╝Ńé»ŃüīŃéĄŃāØŃā╝ŃāłŃüĢŃéīŃü”ŃüäŃüŠŃüÖ | XML | ŃéĄŃéżŃāłŃā×ŃāāŃāŚ | Ńā¢ŃāŁŃé░ | ŃāŚŃā®ŃéżŃāÉŃéĘŃā╝ŃāØŃā¬ŃéĘŃā╝
IPv6ŃāŹŃāāŃāłŃā»Ńā╝Ńé»ŃüīŃéĄŃāØŃā╝ŃāłŃüĢŃéīŃü”ŃüäŃüŠŃüÖ | XML | ŃéĄŃéżŃāłŃā×ŃāāŃāŚ | Ńā¢ŃāŁŃé░ | ŃāŚŃā®ŃéżŃāÉŃéĘŃā╝ŃāØŃā¬ŃéĘŃā╝
õ╝ØĶ©ĆŃéƵ«ŗŃüÖ
Ńé╣ŃéŁŃāŻŃā│ŃüŚŃü”WeChatŃü½ķĆüõ┐Ī :


 English
English fran├¦ais
fran├¦ais čĆčāčüčüą║ąĖą╣
čĆčāčüčüą║ąĖą╣ espa├▒ol
español português
portugu├¬s ž¦┘äž╣ž▒ž©┘Ŗž®
ž¦┘äž╣ž▒ž©┘Ŗž® µŚźµ£¼Ķ¬×
µŚźµ£¼Ķ¬× T├╝rk├¦e
T├╝rk├¦e Óż╣Óż┐ÓżéÓż”ÓźĆ
Óż╣Óż┐ÓżéÓż”ÓźĆ Indonesia
Indonesia 